Pfizer Inc.
NYSE: PFE
Share price (04/01/26): $90.08
Industry: Pharmaceutical Preparations
Financials
|
Type
|
Title
|
Date
|
Extracts
|
|||
|---|---|---|---|---|---|---|
| 10-K | 02/26/26 | PR | IS | BS | CFS | |
| 10-Q | 11/04/25 | PR | IS | BS | CFS | |
| 10-Q | 08/05/25 | PR | IS | BS | CFS | |
| 10-Q | 05/05/25 | PR | IS | BS | CFS | |
| 10-K | 02/27/25 | PR | IS | BS | CFS | |
| 10-Q | 11/04/24 | PR | IS | BS | CFS | |
| 10-Q | 08/05/24 | PR | IS | BS | CFS | |
| 10-Q | 05/08/24 | PR | IS | BS | CFS | |
| 10-K | 02/22/24 | PR | IS | BS | CFS | |
| 10-Q | 11/08/23 | PR | IS | BS | CFS | |
|
|
||||||
Transcripts and slides
|
Title
|
Links
|
Date
|
|
|---|---|---|---|
| Pfizer Inc. Presents at TD Cowen 46th Annual Health Care Conference, Mar-02-2026 10:30 AM | Transcript | 03/02/26 | |
| Earnings Call Q4 FY2025 | Transcript | Slides | 02/03/26 |
| Pfizer Inc. Presents at 44th Annual J.P. Morgan Healthcare Conference, Jan-12-2026 09:45 AM | Transcript | Slides | 01/12/26 |
| Pfizer Inc., 2026 Guidance/Update Call, Dec 16, 2025 | Transcript | 12/16/25 | |
| Pfizer Inc. Presents at Jefferies London Healthcare Conference 2025, Nov-19-2025 09:00 AM | Transcript | 11/19/25 | |
| Pfizer Inc. - Special Call | Transcript | Slides | 11/10/25 |
| Earnings Call Q3 FY2025 | Transcript | Slides | 11/04/25 |
| PFE M&A Announcement | Slides | 09/22/25 | |
| Earnings Call Q2 FY2025 | Transcript | Slides | 08/05/25 |
| Pfizer Inc. - Special Call | Transcript | 06/12/25 | |
|
|
|||
Ownership
|
Type
|
Title
|
Date
|
|---|---|---|
| 4 | 04/01/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| SCHEDULE 13G/A | 03/27/26 | |
| 4 | 03/16/26 | |
| 4 | 03/05/26 | |
|
|
Other
|
Type
|
Title
|
Date
|
|---|---|---|
| ARS | 03/12/26 | |
| SD | 05/30/25 | |
| CERT | 05/20/25 | |
| ARS | 03/13/25 | |
| CORRESP | 08/22/24 | |
| UPLOAD | 08/15/24 | |
| SD | 05/31/24 | |
| ARS | 03/14/24 | |
| ARS | 03/17/23 | |
| UPLOAD | 04/14/22 | |
|
|
News
|
Type
|
Title
|
Date
|
|---|---|---|
| 8-K | 02/03/26 | |
| 8-K | 12/16/25 | |
| 8-K | 11/21/25 | |
| 8-K | 11/13/25 | |
| 8-K | 11/04/25 | |
| 8-K | 08/05/25 | |
| 8-K | 05/19/25 | |
| 8-K | 04/29/25 | |
| 8-K | 04/28/25 | |
| 8-K | 02/04/25 | |
|
|
Prospectuses and Registrations
|
Type
|
Title
|
Date
|
|---|---|---|
| 424B5 | 11/20/25 | |
| FWP | 11/19/25 | |
| 424B3 | 11/18/25 | |
| 8-A12B | 05/19/25 | |
| 424B5 | 05/15/25 | |
| FWP | 05/14/25 | |
| 424B3 | 05/13/25 | |
| POSASR | 05/13/25 | |
| SC TO-I/A | 09/13/24 | |
| SC TO-I/A | 08/22/24 | |
|
|
Proxies
|
Type
|
Title
|
Date
|
|---|---|---|
| DEF 14A | 03/12/26 | |
| DEFA14A | 04/11/25 | |
| DEFA14A | 04/10/25 | |
| DEFA14A | 04/10/25 | |
| DEFA14A | 04/10/25 | |
| DEFA14A | 04/07/25 | |
| PX14A6G | 03/25/25 | |
| DEFA14A | 03/13/25 | |
| DEF 14A | 03/13/25 | |
| PX14A6G | 04/16/24 | |
|
|
Non-Ownership
|
Type
|
Title
|
Filed
|
|---|---|---|
| DEF 14A | 03/12/26 | |
| ARS | 03/12/26 | |
| 10-K | 02/26/26 | |
| 8-K | 02/03/26 | |
| 8-K | 12/16/25 | |
| 8-K | 11/21/25 | |
| 424B5 | 11/20/25 | |
| FWP | 11/19/25 | |
| 424B3 | 11/18/25 | |
| 8-K | 11/13/25 | |
| 10-Q | 11/04/25 | |
| 8-K | 11/04/25 | |
| 8-K | 08/05/25 | |
| 10-Q | 08/05/25 | |
| SD | 05/30/25 | |
| CERT | 05/20/25 | |
| 8-A12B | 05/19/25 | |
| 8-K | 05/19/25 | |
| 424B5 | 05/15/25 | |
| FWP | 05/14/25 | |
|
|
Ownership
|
Type
|
Title
|
Filed
|
|---|---|---|
| 4 | 04/01/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| 4 | 03/30/26 | |
| SCHEDULE 13G/A | 03/27/26 | |
| 4 | 03/16/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
| 4 | 03/05/26 | |
|
|
|
Title
|
Links
|
Date
|
|
|---|---|---|---|
| Pfizer Inc. Presents at TD Cowen 46th Annual Health Care Conference, Mar-02-2026 10:30 AM | Transcript | 03/02/26 | |
| Earnings Call Q4 FY2025 | Transcript | Slides | 02/03/26 |
| Pfizer Inc. Presents at 44th Annual J.P. Morgan Healthcare Conference, Jan-12-2026 09:45 AM | Transcript | Slides | 01/12/26 |
| Pfizer Inc., 2026 Guidance/Update Call, Dec 16, 2025 | Transcript | 12/16/25 | |
| Pfizer Inc. Presents at Jefferies London Healthcare Conference 2025, Nov-19-2025 09:00 AM | Transcript | 11/19/25 | |
| Pfizer Inc. - Special Call | Transcript | Slides | 11/10/25 |
| Earnings Call Q3 FY2025 | Transcript | Slides | 11/04/25 |
| PFE M&A Announcement | Slides | 09/22/25 | |
| Earnings Call Q2 FY2025 | Transcript | Slides | 08/05/25 |
| Pfizer Inc. - Special Call | Transcript | 06/12/25 | |
|
|
|||
BUSINESS

ABOUT PFIZER
Pfizer Inc. is a research-based, global biopharmaceutical company. We apply science and our global resources to bring therapies to people that extend and significantly improve their lives through the discovery, development, manufacture, marketing, sale and distribution of
biopharmaceutical products worldwide. We work across developed and emerging markets to advance wellness, prevention, treatments and cures that challenge the most feared diseases of our time. We collaborate with healthcare providers, governments and local communities to support and expand access to reliable, affordable healthcare around the world. The Company was incorporated under the laws of the State of Delaware on June 2, 1942.
Most of our revenues come from the manufacture and sale of biopharmaceutical products. We believe that our medicines and vaccines provide significant value for healthcare providers and patients through improved treatment of diseases and improvements in health, wellness and productivity as well as by reducing other healthcare costs, such as emergency room visits or hospitalizations. We seek to enhance the value of our medicines and vaccines and actively engage in dialogues about how we can best work with patients, physicians and payors to prevent and treat disease and improve outcomes. We seek to maximize patient access and evaluate our pricing arrangements and contracting methods with payors to minimize adverse impact on our revenues within the current legal and pricing structures.
We are committed to fulfilling our purpose: Breakthroughs that change patients’ lives. Our purpose fuels everything we do and reflects both our passion for science and our commitment to patients. As a science-driven global biopharmaceutical company, we remain focused on advancing our product pipeline, supporting our marketed brands and deploying capital responsibly, with a focus on initiatives that can help contribute to our long-term revenue and future growth.
Our 2026 key priorities are:
1.Maximize value of key transactions
2.Deliver on critical R&D milestones
3.Invest to maximize post-2028 growth
4.Scale AI across our business.
We are committed to strategically capitalizing on growth opportunities, primarily by advancing our own product pipeline and maximizing the value of our existing products, but also through various business development activities. We view our business development activity as an enabler of our strategies and seek to generate growth by pursuing opportunities and transactions that have the potential to strengthen our business and our capabilities. We assess our business, assets and scientific capabilities/portfolio as part of our regular, ongoing portfolio review process and also continue to consider business development activities that will help advance our business strategy.
In addition, we are scaling AI across R&D, manufacturing, commercial and patient engagement to improve productivity and accelerate innovation.
COMMERCIAL OPERATIONS
We manage our commercial operations through a global structure consisting of three operating segments, each led by a single manager: Biopharma, PC1 and Pfizer Ignite. Biopharma, our innovative science-based biopharmaceutical business, is engaged in the discovery, development, manufacture, marketing, sale and distribution of biopharmaceutical products worldwide. PC1 is our contract development and manufacturing organization and a leading supplier of specialty active pharmaceutical ingredients. Pfizer Ignite is an offering that provides strategic guidance and end-to-end R&D services to select innovative biotech companies that align with our R&D focus areas. In 2025, Pfizer made the decision to discontinue Pfizer Ignite and we are winding down this business while collaborating closely with our Ignite partners to ensure continuity and the successful transition of work. Biopharma is the only reportable segment.
Within our Biopharma reportable segment, our commercial divisions market, sell and distribute our products, and global operating functions are responsible for the research, development, manufacturing and supply of our products. The commercial structure within our Biopharma reportable segment in 2025 was composed of the Pfizer U.S. Commercial Division, which focused on the commercialization of Pfizer’s entire product portfolio in the U.S., and the Pfizer International Commercial Division, which focused on the commercialization of Pfizer’s entire product portfolio in all international markets.
As part of our continued focus on commercial execution, at the beginning of 2026, we made changes in our commercial structure, which included the transition of certain off-patent branded and generic sterile injectables and biosimilars from the Specialty Care and Oncology product portfolios to a new Global Hospital and Biosimilars organization within our Biopharma reportable segment. Effective January 1, 2026, the commercial structure within our Biopharma reportable segment is as follows:
Division | Description | ||||
Pfizer U.S. Commercial | Includes the U.S. commercial organization covering Pfizer’s entire product portfolio except for the Global Hospital and Biosimilars organization, as well as the Global Access & Value, Global Chief Marketing Office and Primary Care and Specialty Care U.S. Medical Affairs organizations. | ||||
Pfizer International Commercial | Includes the ex-U.S. commercial and medical affairs organizations covering Pfizer’s entire product portfolio in all international markets except for the Global Hospital and Biosimilars organization in certain international markets. | ||||
Global Hospital and Biosimilars | Includes the commercial organization covering Pfizer’s product portfolio of off-patent branded and generic sterile injectables and biosimilars except in China, Hong Kong and certain other international markets. | ||||
Customer groups and select products within the Biopharma product portfolio in 2026 include:
•Primary Care:
◦Internal medicine product portfolio including in cardiovascular metabolic diseases – select products include: Eliquis, as well as other brands that have experienced patent-based expirations or loss of regulatory exclusivity.
◦Migraine product portfolio: Nurtec ODT/Vydura and Zavzpret.
◦Vaccines product portfolio across all ages – select products include: the Prevnar family, Comirnaty, Abrysvo, FSME/IMMUN-TicoVac, Nimenrix and Trumenba.
◦Treatment for COVID-19: Paxlovid.
•Specialty Care:
◦Inflammation & immunology product portfolio – select products include: Xeljanz, Enbrel (outside the U.S. and Canada), Cibinqo, Litfulo, Eucrisa and Velsipity.
◦Rare disease product portfolio for a number of therapeutic areas with rare diseases, including amyloidosis, hemophilia and endocrine diseases – select products include: the Vyndaqel family, Genotropin, BeneFIX, Xyntha, Somavert, Ngenla and Hympavzi.
◦Certain anti-infective and immunoglobulin medicines – select products include: Zavicefta (outside the U.S. and Canada), Octagam and Panzyga.
•Oncology:
◦Innovative oncology product portfolio of ADCs, small molecules, bispecifics and other immunotherapies that treat a wide range of cancers including certain types of breast cancer, genitourinary cancer and hematologic malignancies, as well as certain types of melanoma, gastrointestinal, gynecological and lung cancer – select products include: Ibrance, Xtandi, Padcev, Adcetris, Inlyta, Lorbrena, Bosulif, Tukysa, Braftovi, Mektovi, Orgovyx, Elrexfio, Tivdak and Talzenna.
•Hospital and Biosimilars:
◦Product portfolio of off-patent branded and generic sterile injectables, oncology biosimilars and biosimilars for chronic immune and inflammatory diseases – select products include: Biosimilars – Inflectra, Oncology biosimilars such as Retacrit, Ruxience, Zirabev, Trazimera and Nivestym, and other biosimilars; and Sterile Injectables – Sulperazon (outside the U.S. and Canada), Atgam, Fragmin, Solu Medrol, Solu Cortef and Bicillin.
RESEARCH AND DEVELOPMENT
R&D is at the heart of fulfilling our purpose to deliver breakthroughs that change patients’ lives as we work to translate advanced science and technologies into the medicines and vaccines that may be the most impactful for patients. In addition to discovering and developing new products, our R&D efforts seek to add value to our existing products by improving their safety, efficacy and ease of dosing and by discovering potential new indications.
Our R&D Priorities and Strategy. Our R&D priorities include:
•delivering a pipeline of highly differentiated medicines and vaccines where we have a unique opportunity to bring the most important new therapies to patients in need;
•advancing our capabilities that can position us for long-term R&D leadership; and
•advancing new models for partnerships with creativity, flexibility and urgency to deliver innovation to patients as quickly as possible.
To that end, our R&D primarily focuses on our main therapeutic areas, which are oncology, internal medicine (including cardiometabolic, weight management and migraine), vaccines (with a pipeline focus on infectious diseases with significant unmet medical need) and inflammation and immunology.
While a significant portion of our R&D is internal, we also seek promising chemical and biological lead molecules and innovative technologies developed by others to incorporate into our discovery and development processes or projects, as well as our portfolio. We do so by entering into collaboration, alliance and license agreements with universities, biotechnology companies and other firms as well as through acquisitions and investments, including co-funding agreements with third-parties. These arrangements allow us to share knowledge, risk and cost. They also enable us to access external scientific and technological expertise, as well as provide us the opportunity to advance our own products and in-licensed or acquired products.
Our R&D Operations. In 2025, we continued to enhance our global R&D operations and pursued strategies to improve R&D productivity and advance a sustainable and value-creating pipeline. We manage our R&D operations for all therapeutic areas in a single R&D organization led by the Chief Scientific Officer and President, Research and Development. This organization is responsible for overseeing all R&D activities with end-to-end responsibilities that span from discovery to late-phase clinical development and post-approval activities, including facilitating regulatory submissions, engaging with health authorities and global medical strategies, as well as U.S. Oncology medical affairs. We continue to evaluate how our simplified structure and sharpened focus might lead to improvements in productivity and potential efficiencies. For example, approximately $500 million of R&D savings achieved from our ongoing realigning our cost base program in 2025 is expected to be reinvested in R&D programs in 2026. See the Costs and Expenses––Restructuring Charges and Other Costs Associated with Acquisitions and Cost-Reduction/Productivity Initiatives section within MD&A for more information.
We manage R&D operations on a total-company basis through the organization described above. The Portfolio Management Team (PMT), chaired by our Chief Strategy and Innovation Officer, Executive Vice President, is accountable for aligning resources across R&D, and for helping to ensure optimal capital allocation across the R&D portfolio. We believe that this approach also serves to maximize accountability and flexibility.
We do not disaggregate total R&D expense by development phase or by therapeutic area since, as described above, we manage our R&D strategy and operations collectively under the governance of the PMT and do not manage our R&D operational spend independently by development phase or by therapeutic area. Further, as we are able to adjust a significant portion of our spending quickly, we believe that any prior-period information about R&D expense by development phase or by therapeutic area would not necessarily be representative of future spending.
Our R&D Pipeline. The process of drug, vaccine and biological product discovery from initiation through development and to potential regulatory approval is lengthy and can take more than ten years. As of February 3, 2026, we had the following number of projects in various stages of R&D:
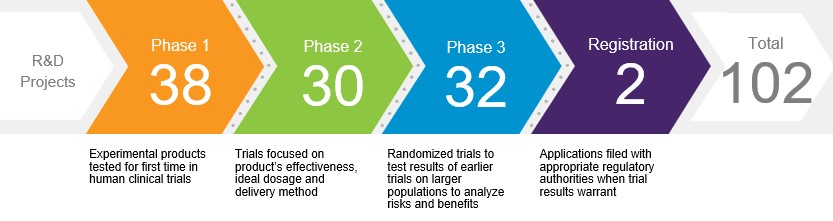
Development of a single compound is often pursued as part of multiple programs. While our product candidates may or may not receive regulatory approval, new candidates entering clinical development phases are the foundation for future products. Information concerning several of our drug, vaccine and biological candidates in development, as well as supplemental filings for existing products, is set forth in the Product Developments section within MD&A. The discovery and development of drugs, vaccines and biological products are time consuming, costly and unpredictable.
COLLABORATION AND CO-PROMOTION AGREEMENTS
We use collaboration and/or co-promotion arrangements to enhance our R&D, sales and distribution of certain biopharmaceutical products, which include, among others, the following:
•Comirnaty is an mRNA-based coronavirus vaccine to help prevent COVID-19, which is jointly developed and commercialized with BioNTech. Pfizer and BioNTech equally share the costs of development for the Comirnaty program. We also share gross profits equally from commercialization of Comirnaty (excluding mainland China, Hong Kong, Macau and Taiwan, where we do not have rights), subject to regulatory authorizations or approvals market by market. For discussion on Comirnaty, see the Overview of Our Performance, Operating Environment, Strategy and Outlook—COVID-19 section within MD&A.
•Eliquis (apixaban) is part of the novel oral anticoagulant market and was jointly developed and is commercialized with BMS as an alternative treatment option to warfarin in appropriate patients. We fund between 50% and 60% of all development costs depending on the study, and profits and losses are shared equally except in certain countries where we commercialize Eliquis and pay a percentage of net sales to BMS. In certain smaller markets we have full commercialization rights and BMS supplies the product to us at cost plus a percentage of the net sales to end-customers.
•Xtandi (enzalutamide) is an androgen receptor inhibitor that blocks multiple steps in the androgen receptor signaling pathway within tumor cells that is being developed and commercialized in collaboration with Astellas, which has exclusive commercialization rights for Xtandi outside the U.S. We share equally in the gross profits and losses related to U.S. net sales and also share equally all Xtandi commercialization costs attributable to the U.S. market, subject to certain exceptions. In addition, we share certain development and other collaboration expenses. For international net sales we receive royalties based on a tiered percentage.
•Orgovyx (relugolix) is an oral gonadotropin-releasing hormone (GnRH) receptor antagonist for the treatment of adult patients with advanced prostate cancer that is being developed and commercialized with SMPS. The companies equally share profits and allowable expenses in the U.S. for Orgovyx. Pfizer does not have rights outside of this market. Separately, in December 2024, the companies terminated their collaboration with respect to the relugolix combination tablet.
•Padcev (enfortumab vedotin-ejfv) is a first-in-class ADC that is directed to Nectin-4, a protein located on the surface of cells and highly expressed in bladder cancer, that is being co-developed and jointly commercialized with Astellas. In the U.S., Padcev has been approved for use with pembrolizumab for adult patients with locally advanced or metastatic urothelial cancer and for adult patients with muscle-invasive bladder cancer (MIBC) who are ineligible for cisplatin-containing chemotherapy. Other approvals and indications for Padcev vary by market. In the U.S., Pfizer and Astellas jointly promote, and we record net sales and are responsible for all U.S. distribution activities for Padcev. The companies each bear the costs of their own sales organizations in the U.S., and equally share certain other costs associated with commercializing and any profits realized for Padcev in the U.S. Outside the U.S., we have commercialization rights in all countries in North and South America, and Astellas has commercialization rights in the rest of the world. The agreement between us and Astellas provides that the companies will effectively equally share in profits realized in markets outside of the U.S. through: (i) a costs-incurred and profit-sharing mechanism based on product sales and costs of commercialization in certain markets and (ii) a royalty-payment mechanism intended to approximate an equal profit share for both parties in the remaining markets.
•Adcetris (brentuximab vedotin) is being developed and commercialized in collaboration with Takeda. Pfizer has commercialization rights for Adcetris in the U.S. and its territories and in Canada. Takeda has commercialization rights in the rest of the world and pays Pfizer a royalty based on a percentage of Takeda’s net sales of Adcetris in its licensed territories, based on annual net sales tiers.
•Tivdak (tisotumab vedotin-tftv) is commercialized in collaboration with Genmab. Pfizer has co-promotion rights in the U.S. Outside the U.S., Genmab has the sole right to promote Tivdak for second-line plus mCC and has co-promotion rights for other indications in all territories except certain territories where Zai Lab Limited (Zai Lab) has commercialization rights (mainland China, Hong Kong, Macau, and Taiwan). Our profit sharing rights, royalty rights and expense obligations relating to Tivdak vary by jurisdiction.
In addition, we have collaboration and/or co-promotion arrangements with respect to certain other biopharmaceutical products.
Revenues associated with these arrangements are included in Alliance revenues (except in certain markets where we have direct sales and except for the majority of revenues for Comirnaty and Padcev, which are included in Product revenues). Business—Patents and Other Intellectual Property Rights section, the Item 1A. Risk Factors—Collaborations and Other Relationships with Third Parties section and Notes 2 and 17.
INTERNATIONAL OPERATIONS
Our operations are conducted globally, and we supply our medicines and vaccines to approximately 200 countries and territories. Emerging markets are an important component of our strategy for global leadership, and our commercial structure recognizes that the demographics and rising economic power of the fastest-growing emerging markets are becoming more closely aligned with the profile found within developed markets. Urbanization and the rise of the middle class in emerging markets provide potential growth opportunities for our products.
Revenues from operations outside the U.S. of $25.5 billion, $24.9 billion and $31.4 billion accounted for 41%, 39% and 53% of Total revenues in 2025, 2024 and 2023, respectively. Revenues exceeded $500 million in each of 12, 11 and 14 countries outside the U.S. in 2025, 2024 and 2023, respectively. As a percentage of Total revenues, China was our largest market outside the U.S. in 2025 and 2024 (representing 5% and 4% of total revenues, respectively), and Japan was our largest market in 2023 (representing 6% of total revenues). For a geographic breakdown of Total revenues
Our international operations are subject to risks inherent in carrying on business in other countries. Business—Government Regulation and Price Constraints sections.
SALES AND MARKETING
Our prescription biopharmaceutical products are sold principally to wholesalers, but we also sell directly to retailers, hospitals, clinics, government agencies and pharmacies. Our vaccines in the U.S. are primarily sold directly to the federal government (including the CDC), wholesalers, individual provider offices, retail pharmacies and integrated delivery systems. Our vaccines outside the U.S. are primarily sold to government and non-government institutions. Certain of these government contracts may be renegotiated or terminated at the discretion of a government entity.
We also seek to gain access for our products on formularies, which are lists of approved medicines available to members of healthcare programs or PBMs in the U.S. Insurers and PBMs who design and negotiate formularies on their behalf use various benefit designs, such as tiered co-pays for formulary products, to drive utilization of products in preferred formulary positions, typically in exchange for a discount off the price of the medicine in the form of a rebate agreement. We may also work with payors on disease management programs that help to develop tools and materials to educate patients and physicians on key disease areas.
We promote our products to healthcare providers and patients consistent with applicable laws, regulations and policies. Through our marketing organizations, we explain the approved uses, benefits and risks of our products to healthcare providers and patients and, in the U.S., to MCOs that provide insurance coverage, such as hospitals, integrated delivery systems, PBMs and health plans; and employers and government agencies who hire MCOs to provide health benefits to their employees. In the U.S. and select international markets, we market directly to consumers through direct-to-consumer advertising that seeks to communicate the approved uses, benefits and risks of our products while motivating people to have meaningful conversations with their doctors. In addition, we sponsor general advertising to educate the public on disease awareness, prevention and wellness, important public health issues and our patient assistance programs. Further, pursuant to our voluntary agreement with the U.S. government, we are participating on the TrumpRx.gov platform, which allows U.S. patients to purchase certain medicines at significant discounts to current retail prices.
As part of our commitment to engaging our customers in a manner they prefer, we take an omnichannel approach, including both virtual and in person interactions, and see generally positive customer response to both approaches.
PATENTS AND OTHER INTELLECTUAL PROPERTY RIGHTS
Patents. We own or have co-promotion and/or license rights related to a number of patents covering pharmaceutical and other products, their uses, formulations, and product manufacturing processes.
Patents for individual products extend for varying periods according to the date of patent filing or grant and the legal term of patents in the various countries where patent protection is obtained. The scope of protection afforded by a patent can vary from country to country and depends on the patent type, the scope of its patent claims and the availability of legal remedies. Patent term extensions (PTE) may be available in some countries to compensate for a loss of patent term due to delay in a product’s approval due to the regulatory requirements, while patent term adjustment may be available in some countries to compensate for administrative delays during prosecution of patents. One of the primary considerations in limiting our operations in some countries outside the U.S. is the lack of effective intellectual property protection for our products, although international and U.S. free trade agreements have included some global protection of intellectual property rights. Business—Government Regulation and Price Constraints section.
In various markets, a period of regulatory exclusivity may be provided for drugs or vaccines upon approval. The scope and term of such exclusivity will vary but, in general, the period will run concurrently with the term of any existing patent rights associated with the drug at the time of approval.
Based on current sales and other factors, and considering the competition with products sold by our competitors, the patent rights we consider most significant in relation to our business as a whole, together with the year in which the basic product patent expires, are as follows:
| Product | U.S. Basic Product Patent Expiration Year(1) | Major Europe Basic Product Patent Expiration Year(1) | Japan Basic Product Patent Expiration Year(1) | |||||||||||||||||
| Xeljanz | 2026 | 2028(2) | 2025 | |||||||||||||||||
| Prevnar 13/Prevenar 13 | 2026 | (3) | 2029 | |||||||||||||||||
Adcetris(4) | 2026 | (4) | (4) | |||||||||||||||||
Eliquis | 2027(5) | 2026(6) | 2026 | |||||||||||||||||
| Ibrance | 2027 | 2028 | 2028 | |||||||||||||||||
Xtandi(7) | 2027 | (7) | (7) | |||||||||||||||||
| Vyndaqel/Vyndamax/Vynmac | 2026 (2028 pending PTE)(8) | 2026 | 2026/2029(9) | |||||||||||||||||
Xalkori | 2029 | 2027(10) | 2028 | |||||||||||||||||
| Product | U.S. Basic Product Patent Expiration Year(1) | Major Europe Basic Product Patent Expiration Year(1) | Japan Basic Product Patent Expiration Year(1) | |||||||||||||||||
Braftovi(11) | 2030 (2031 pending PTE) | (11) | (11) | |||||||||||||||||
Mektovi(11) | 2026 (2027 pending PTE)(12) | (11) | (11) | |||||||||||||||||
Talzenna | 2029 (2032 pending PTE) | 2034 | 2034 | |||||||||||||||||
| Lorbrena | 2033 | 2034 | 2036 | |||||||||||||||||
Padcev(13) | 2033 | (13) | (13) | |||||||||||||||||
Tukysa | 2031 (2034 pending PTE) | 2031 | 2034(2) | |||||||||||||||||
| Zavzpret | 2031 (2034 pending PTE) | 2031(14) | 2031(14) | |||||||||||||||||
| Velsipity | 2030 (2035 pending PTE) | 2034 | 2035(2) | |||||||||||||||||
Prevnar 20/Prevenar 20 | 2035 | 2037 | 2038 | |||||||||||||||||
| Nurtec ODT/Vydura | 2034 | 2035 | 2035(2) | |||||||||||||||||
Ngenla(15) | 2035(2) | 2032(2) | 2030(2) | |||||||||||||||||
| Cibinqo | 2036 | 2036 | 2038 | |||||||||||||||||
Tivdak(16) | 2035 | (16) | (16) | |||||||||||||||||
| Litfulo | 2034 (2037 pending PTE) | 2038 | 2039 | |||||||||||||||||
| Abrysvo | 2036 (2037 pending PTE) | 2036 | 2036 (2039 pending PTE) | |||||||||||||||||
Elrexfio | 2036 (2037 pending PTE) | 2036 (2038 pending SPC) | 2036 (2038 pending PTE) | |||||||||||||||||
Hympavzi | 2036 (2038 pending PTE) | (17) | 2036 (2041 pending PTE) | |||||||||||||||||
Comirnaty(18) | 2041 | (17)(19) | 2041 | |||||||||||||||||
| Paxlovid | 2041 | 2041 | 2041 | |||||||||||||||||
(1)Unless otherwise indicated, the years pertain to the basic product patent expiration, including granted PTEs, supplementary protection certificates (SPC) or pediatric exclusivity periods. SPCs are included when granted in three out of five major European markets (France, Germany, Italy, Spain and the U.K.). Noted in parentheses is the projected year of expiry of the earliest pending patent term extension in the U.S. or Japan and/or SPC application in Europe, the term of which, if granted, may be shorter than originally requested due to a number of factors. In some instances, there are later-expiring patents relating to our products which may or may not protect our product from generic or biosimilar competition after the expiration of the basic patent.
(2)Expiry is provided by regulatory exclusivity in this market.
(3)The Europe patent that covers the combination of the 13 serotype conjugates of Prevenar 13 was revoked following an opposition and has now been withdrawn. There are other Europe patents and pending applications covering the formulation, various aspects of the manufacturing process, and the combination of serotype conjugates of Prevenar 13 that remain in force.
(4)Adcetris is commercialized in collaboration with Takeda. Pfizer has commercialization rights for Adcetris in the U.S. and its territories and in Canada. Takeda has commercialization rights in the rest of the world.
(5)Eliquis was jointly developed and is commercialized in collaboration with BMS. In the U.S., we and BMS previously settled certain patent litigations with a number of generic companies permitting their launch of a generic version of Eliquis on April 1, 2028 (the settled generic companies). We continued to litigate against three remaining generic companies and following the resolution of the litigation in our favor, the three generic companies are not permitted to launch their products until the 2031 expiration date of the formulation patent. Both the composition of matter patent expiring in November 2026 and the formulation patent expiring in 2031 may be subject to future challenges. While we cannot predict the outcome of any potential future litigation, there are certain potential alternatives that might occur which could potentially permit generic launch prior to April 1, 2028: (i) if the formulation patent is held invalid or not infringed in future litigation, through appeal, the settled generic companies and any successful future litigant would be permitted to launch on November 21, 2026; or (ii) if both patents are held invalid or not infringed in future litigation, through appeal, the settled generic companies and any successful future litigant could launch products immediately upon such an adverse decision.
(6)The apixaban basic product patent and associated SPC were invalidated in the U.K. Additional challenges are pending in other jurisdictions.
(7)Xtandi is being developed and commercialized in collaboration with Astellas, which has exclusive commercialization rights for Xtandi outside the U.S.
(8)Interim patent term extension requests have been granted extending the expiry from December 2025 to December 2026, and Pfizer has pending applications for patent term extension to 2028.
(9)Vyndaqel (tafamidis meglumine) basic patent expiry in Japan is August 2026 for treatment of polyneuropathy. Vynmac (tafamidis) was approved in Japan for treatment of cardiomyopathy with regulatory exclusivity expiring in March 2029.
(10)Pediatric extension applications have been filed on SPCs for Xalkori in Europe. In France, Germany, and Italy the pediatric extension has been granted, extending the SPC to 2028.
(11)Pfizer has exclusive rights to Braftovi and Mektovi in the U.S., Canada, Latin America, the Middle East and Africa. Ono has exclusive rights to commercialize the product in Japan and South Korea, Medison Pharma Ltd. has exclusive rights to commercialize the product in Israel and Pierre Fabre has exclusive rights to commercialize the product in all other countries, including Europe and Asia (excluding Japan and South Korea). Pfizer receives royalties from Pierre Fabre and Ono on sales of Braftovi and Mektovi.
(12)Mektovi U.S. expiry is provided by a composition of matter patent. Interim patent term extension requests have been filed to extend the expiry from March 2026 to March 2027, and Pfizer has filed an application for patent term extension to August 2027.
(13)Padcev is being commercialized in collaboration with Astellas. Pfizer has co-promotion rights in the U.S. Outside the U.S., Pfizer has commercialization rights in all countries in North and South America, and Astellas has commercialization rights in the rest of the world, including Europe, Asia, Australia and Africa.
(14)Product not yet approved or authorized in this market.
(15)Ngenla is licensed from OPKO Health, Inc., and is developed and commercialized by Pfizer, including in the U.S., Latin America, Europe, Africa, and Asia.
(16)Tivdak is commercialized in collaboration with Genmab. Pfizer has co-promotion rights in the U.S. Outside the U.S., Genmab has the sole right to promote Tivdak for second-line plus mCC and has co-promotion rights for other indications in all territories except certain territories where Zai Lab has commercialization rights (mainland China, Hong Kong, Macau, and Taiwan).
(17)The basic product patent application has been filed in this market. If granted, a full term is expected in this market.
(18)Product is being commercialized in collaboration with BioNTech. The Comirnaty trademark covers marketed variants.
(19)Pfizer does not have co-promotion rights for this product in Germany.
For information regarding past reported, including recently expired, patent expiration dates, please see the Patents and Intellectual Property Rights sections of our prior Annual Reports on Form 10-K. Business—Collaboration and Co-Promotion Agreements.
Loss of Intellectual Property Rights. The loss, expiration or invalidation of intellectual property rights, patent litigation settlements and judgments and the expiration of co-promotion and licensing rights can have a material adverse effect on our revenues. Once patent protection has expired or has been lost prior to the expiration date as a result of a legal challenge, we typically lose market exclusivity on these products, and generic and biosimilar pharmaceutical manufacturers generally produce identical or highly similar products and sell them for a lower price. The date at which generic or biosimilar competition commences may be different from the date that the patent or regulatory exclusivity expires. However, when generic or biosimilar competition does commence, the resulting price competition can substantially decrease our revenues for the impacted products, often in a very short period of time. Also, if one of our product-related patents is found to be invalid by judicial, court, regulatory or administrative proceedings, generic or biosimilar products could be introduced, resulting in the erosion of sales of our existing products. Additionally, we could be subject to claims that our intellectual property rights infringe third party patents.
Certain of our products have experienced patent-based expirations or loss of regulatory exclusivity in certain markets in the last few years, and we expect certain products to face new or increased generic competition over the next few years. We anticipate a significant reduction of revenue from patent-based or regulatory exclusivity expiries in 2026 through 2030 as several of our in-line products experience these expirations, with the rate of the reduction of revenues from patent-based or regulatory exclusivity expiries expected to significantly accelerate over the next few years. There is no assurance that a particular product will maintain market exclusivity for the full time period that appears in the estimates included in this Form 10-K or that we assume when we provide our financial guidance. For additional information on the impact of loss of patent-based exclusivity or regulatory exclusivity on our revenues, see the Overview of Our Performance, Operating Environment, Strategy and Outlook—Our 2025 Performance and —Intellectual Property Rights and Collaboration/Licensing Rights sections within MD&A.
We continue to vigorously defend our patent rights against infringement, and we will continue to support efforts that strengthen worldwide recognition of patent rights while taking necessary steps to help ensure appropriate patient access. See the Item 1A. Risk Factors—Competitive Products, —Intellectual Property Protection and —Third-Party Intellectual Property Claims sections and Note 16A1.
Trademarks. Our products are sold under brand-name and logo trademarks and trade dress. Registrations generally are for fixed, but renewable, terms and protection is provided in some countries for as long as the mark is used while in others, for as long as it is registered. Protecting our trademarks is of material importance to us.
COMPETITION
Our business is conducted in intensely competitive and highly regulated markets. Many of our products face competition in the form of branded or generic drugs or biosimilars that treat similar diseases or indications. The principal forms of competition include efficacy, safety, tolerability, ease of use and cost. Though the means of competition vary among our products, demonstrating the value of our products is a critical factor for success.
We compete with other companies that manufacture and sell products that treat or prevent diseases or indications similar to those treated or prevented by our major products. These competitors include other worldwide research-based biopharmaceutical companies, smaller research companies with more limited therapeutic focus and generic drug and biosimilar manufacturers. Our competitors also may devote substantial funds and resources to R&D and their successful R&D could result in erosion of the sales of our existing products and potential sales of our products in development, as well as product obsolescence. In addition, several of our competitors operate without large R&D expenses and make a regular practice of challenging our product patents before their expiration.
To help address competitive trends we continually emphasize innovation, which is underscored by our multi-billion-dollar investment in R&D, as well as our business development transactions, both designed to result in a strong and differentiated product pipeline. Our investment in research continues even after drug or vaccine approval as we seek to further demonstrate the value of our products for the conditions they treat or prevent, as well as investigating potential new applications. We educate patients, physicians, payors and global health authorities on the benefits and risks of our medicines and vaccines, and seek to continually enhance the organizational effectiveness of our biopharmaceutical functions, including our efforts to effectively launch and market our products to our customers.
Operating conditions have also shifted as a result of increased global competitive pressures, industry regulation and cost containment. We continue to evaluate, adapt and improve our organization and business practices in an effort to better meet customer and public needs. We also continue to support programs to help address patient affordability and access barriers, as we strive to advance fundamental health system change through our support for better healthcare solutions. For example, our Accord for a Healthier World program aims to provide our full portfolio of patented and off-patent medicines and vaccines for which Pfizer holds global rights on a not-for-profit basis to 1.2 billion people living in 45 lower-income countries around the world.
Our vaccines have and may continue to face competition, including from the introduction of alternative vaccines or “next-generation” vaccines prior to or after the expiration of their patents, which may adversely affect our future results.
Our biosimilars, which include biosimilars of certain inflammation & immunology and oncology biologic medicines, compete with branded products from competitors, as well as other generics and biosimilars manufacturers. We seek to maximize the opportunity to establish a “first-to-
market” or early market position for our biosimilars to provide customers a lower-cost alternative as soon as practicable and also to potentially provide us with higher levels of sales and profitability until other competitors enter the market.
Generic Products. Generic pharmaceutical manufacturers pose one of the biggest competitive challenges to our branded small molecule products because they can market a competing version of our product after the expiration or loss of our patent protection, or exclusivity, and often charge much less. Several competitors regularly challenge our product patents before their expiration. Generic competitors often operate without large R&D expenses, as well as without costs of conveying medical information about products to the medical community. In addition, the approval process in the U.S. and in the EU exempts most generics from costly and time-consuming clinical trials to demonstrate their safety and efficacy, allowing generic manufacturers to rely on the safety and efficacy data of the innovator product. In China, for example, given the expansion of the QCE process and continuation of the VBP program, we expect to continue to face intensified competition by certain generic manufacturers in 2026 and beyond, which has and may continue to result in price cuts and volume loss of some of our products. In addition, generic versions of competitors’ branded products have and may continue to compete with our products.
Commercial and government payors typically encourage the use of generics as alternatives to brand-name drugs in their healthcare programs, including Medicaid in the U.S., and U.S. laws generally allow, and in some cases require, pharmacists to substitute generic drugs for brand-name drugs. In a small subset of states, prescribing physicians are able to expressly prevent such substitution. Similar rules also apply in several EU member states, where national authorities typically encourage and incentivize the use of generic products.
Biosimilars. Certain of our biologic products, including Enbrel (we market Enbrel outside the U.S. and Canada), already face, or may face in the
future, competition from biosimilars (also referred to as follow-on biologics). Biosimilars are versions of biologic medicines that have been developed and proven to be highly similar to the original biologic in terms of safety and efficacy and that have no clinically meaningful differences in safety, purity or potency. Biosimilars have the potential to offer high-quality, lower-cost alternatives to innovative biologic medicines. In the U.S., biosimilars referencing innovative biologic products are approved by the FDA under the U.S. Public Health Service Act, whereas in the EU the EMA is responsible for evaluating the majority of applications for biosimilars through the centralized procedure.
PRICING PRESSURES AND MANAGED CARE ORGANIZATIONS
Commercial Pricing Pressures. Pricing and access pressures in the commercial sector continue to be significant. Overall, increasing pressure exists on U.S. providers to deliver healthcare at a lower cost and to ensure that those expenditures deliver demonstrated value in terms of health outcomes. Many employers have adopted or make available high deductible health plans, which can increase out-of-pocket costs for medicines, or are using utilization management tools or limiting access on formularies. This trend is likely to continue. Private third-party payors, such as health plans, increasingly challenge pharmaceutical product pricing, which could result in lower prices, lower reimbursement rates for payors and a reduction in demand for our products, including denial of coverage of our products, if lower cost alternatives are available. Payors often require significant discounts, or rebates, from our prices in exchange for more favorable formulary placement. Pricing pressures also may occur as a result of highly competitive biopharmaceutical markets and increasing concentration of insurers and PBMs. Healthcare provider purchasers, directly or through group purchasing organizations, are seeking enhanced discounts or implementing more rigorous bidding or purchasing review processes.
We believe medicines and vaccines are the most efficient and effective use of healthcare dollars based on the value they deliver to the overall healthcare system. We work with law makers and advocate for solutions that effectively improve patient health outcomes, lower costs to the healthcare system, and help ensure access to medicines and vaccines within an efficient and affordable healthcare system. This includes assessing our go-to market model to help address patient affordability challenges. We have engaged with major payors and the U.S. government to explore opportunities to improve access and reimbursement in an effort to drive pro-patient policies. In addition, in response to the evolving U.S. and global healthcare spending landscape, we work with health authorities, health technology assessment and quality measurement bodies and major U.S. payors throughout the product-development process to better understand how these entities value our compounds and products. Further, we are developing stronger support designed to demonstrate the value of the medicines and vaccines that we discover or develop, register and manufacture.
Business—Government Regulation and Price Constraints and Item 1A. Risk Factors—Pricing and Reimbursement sections.
Managed Care Organizations. The evolution of managed care in the U.S. has been a major factor in the competitiveness of the healthcare marketplace. Approximately 314 million people in the U.S. now have some form of health insurance coverage, and the marketing of prescription drugs and vaccines to both consumers and the entities that manage coverage in the U.S. continues to grow in importance. In particular, the influence of MCOs has increased in recent years due to the growing number of patients receiving coverage through MCOs. At the same time, consolidation in the MCO industry has resulted in fewer, even larger MCOs, which enhances those MCOs’ ability to negotiate lower pricing and further increases their importance to our business. Since MCOs purport to seek to contain and reduce healthcare expenditures, their growing influence has increased downward pressure on drug prices, as well as negatively impacted revenues.
MCOs and their PBMs typically negotiate prices with pharmaceutical providers by using formularies (which are lists of approved medicines available to MCO members), clinical protocols (which require prior authorization for a branded product if a generic product is available or require the patient to first fail on one or more generic products before permitting access to a branded medicine), long-term contracts and their ability to influence volume and market share of prescription drugs. In addition, by placing branded medicines on higher-tier or non-preferred status in their formularies, MCOs transfer to the patient higher patient out-of-pocket expenses. This financial disincentive is a tool for MCOs to manage drug costs and channel patients to medicines preferred by the MCOs. We expect payment reforms for MCOs will continue to evolve with increased emphasis on expanded participation and on removing barriers to equitable healthcare.
The breadth of the products covered by formularies can vary considerably from one MCO to another, and many formularies include alternative and competitive products for treatment of particular medical problems. MCOs emphasize primary and preventive care, out-patient treatment and procedures performed at doctors’ offices and clinics as ways to manage costs. Hospitalization and surgery, typically the most expensive forms of treatment, are carefully managed, and drugs that can help in chronic care management and reduce the need for hospitalization, professional therapy or surgery may become favored first-line treatments for certain diseases. At the same time, MCOs may seek to exclude high-cost drugs from formularies in their efforts to manage and lower their costs.
Exclusion of a product from a formulary or other restrictions can significantly impact drug usage in the MCO or PBM managed patient population and beyond. Consequently, pharmaceutical companies compete to gain access to formularies for their products, typically on the basis of unique product features, such as greater efficacy, better patient ease of use, or fewer side effects, as well as the overall cost of the therapy. We continue
to seek to ensure that our major products are included on MCO formularies. However, our branded products are increasingly being placed on the higher tiers or in a non-preferred status. Continuing efforts by managed care entities to contain or reduce costs of healthcare and/or impose price controls may adversely affect demand for our products and our financial performance.
Over the last year, PBM practices have come under scrutiny from Federal and State policymakers. These legislations, enforcements, settlements and related guidance and rules could have implications to our business, including how we engage with these entities as well as the formulary status of our products.
Agreement with the U.S. Government. In September 2025, we announced an agreement with the Trump Administration in which we voluntarily agreed to implement measures designed to make certain drug prices for U.S. patients more comparable to those in other developed countries. We are also participating in the TrumpRx.gov platform, which allows U.S. patients to purchase certain medicines at significant discounts to current retail prices, where the large majority of the Company’s primary care treatments and some select specialty brands will be offered at savings that will range as high as 85% and on average 50%. The September 2025 agreement also provides a three-year grace period during which time our products will not face Section 232 tariffs, provided the Company further invests in manufacturing in the U.S. Pfizer is now in the process of entering into binding final agreements to implement these arrangements. Business—Government Regulation and Price Constraints and Item 1A. Risk Factors—Pricing and Reimbursement sections.
RAW MATERIALS
We procure raw materials essential to our business from numerous suppliers worldwide. In general, these materials have been available in sufficient quantities to support our demand and in many cases are available from multiple suppliers. We do not anticipate the availability of raw materials to have a significant impact on our operations in 2026, but are monitoring potential supply chain disruptions as a result of ongoing geopolitical and trade negotiations, which could, among other things, impact costs. We are continuing to monitor and implement mitigation strategies to reduce any potential risk or impact including active supplier management, qualification of additional suppliers and advanced purchasing to the extent possible.
GOVERNMENT REGULATION AND PRICE CONSTRAINTS
We are subject to extensive regulation by government authorities in the countries in which we do business. This includes laws and regulations governing the operations of biopharmaceutical companies, such as the research and development, testing, approval, manufacturing and marketing of products, pricing (including discounts and rebates) and price reporting, interactions with healthcare professionals, institutions, and referral sources, reporting of remuneration provided to healthcare providers and academic medical centers, financial assistance provided to patients, clinical research, data privacy and information security, among others. These laws and regulations may require administrative guidance for implementation, and a failure to comply could subject us to legal and/or administrative actions. Enforcement measures may include substantial fines and/or penalties, orders to stop non-compliant activities, criminal charges, warning letters, product recalls or seizures, delays in product approvals, exclusion from participation in government programs or contracts as well as limitations on conducting business in applicable jurisdictions, and could result in harm to our reputation and business. Compliance with these laws and regulations is costly, and requires significant technical expertise and capital investment to ensure compliance.
In the U.S.
Drug and Biologic Regulation. The FDA, pursuant to the FFDCA, the Public Health Service Act and other federal statutes and regulations, extensively regulates pre- and post-marketing activities related to our biopharmaceutical products and devices. The statutes and regulations govern areas such as safety and efficacy, clinical trials, advertising and promotion, quality control, manufacturing, labeling, distribution, post-marketing safety surveillance and reporting, and record keeping. Other U.S. federal agencies, including the DEA, may also regulate certain of our products and activities.
For a biopharmaceutical company to market a drug or a biologic product, including vaccines, the FDA must approve a product’s NDA or BLA (or supplemental NDA or supplemental BLA). Prior to such approval, the FDA will evaluate whether the product is safe and effective for its intended use.
A drug or biologic may be subject to post marketing commitments, which are studies or clinical trials that the product sponsor agrees to conduct, or post marketing requirements, which are studies or clinical trials that are required as a condition of approval. In addition, we are also required to report adverse events and comply with cGMPs (the FDA regulations that govern all aspects of manufacturing quality for pharmaceuticals) and the Drug Supply Chain Security Act (the law that, among other things, sets forth requirements related to product tracing, product identifiers and verification for manufacturers, wholesale distributors, third-party logistics providers, re-packagers and dispensers to facilitate the tracing of product through the pharmaceutical distribution supply chain), as well as advertising and promotion regulations. See the Item 1A. Risk Factors—Development, Regulatory Approval and Marketing of Products and —Post-Authorization/Approval Data sections. We are also responsible for monitoring, reviewing, and the periodic reporting of adverse drug experience, or pharmacovigilance, including information received from any source, such as commercial marketing experience, postmarketing clinical investigations, postmarketing epidemiological or surveillance studies, clinical trials conducted by other parties within and outside the U.S., reports in the scientific literature, and unpublished scientific papers.
In the context of public health emergencies, like the COVID-19 pandemic, we may apply to the FDA for an EUA which, if granted, allows for the distribution and use of our products during the declared emergency, in accordance with the conditions set forth in the EUA, unless the EUA is terminated by the government. Although the criteria for an EUA differ from the criteria for approval of an NDA or BLA, EUAs nevertheless require the development and submission of data to satisfy the relevant FDA standards, and a number of ongoing obligations. The FDA generally expects EUA holders to work toward submission of full applications, such as a BLA or an NDA, as soon as possible.
Biosimilar Regulation. The FDA regulates biosimilars which are follow-on products that reference an innovative biological product. A product is biosimilar if the products are highly similar and there are no clinically meaningful differences between them. A biosimilar is interchangeable if switching the products does not decrease safety or efficacy. In many states, interchangeable biosimilars may be substituted for the reference product at the pharmacy. A biosimilar application may not be filed until four years, and not approved until 12 years, after reference product licensure. No other interchangeable may be approved until one year after approval of the first interchangeable.
Sales and Marketing Regulations. Our marketing and promotional practices are subject to federal and state laws, such as the Anti-Kickback Statute (AKS), Civil Monetary Penalties Law, False Claims Act and state laws governing kickbacks and false claims, intended to prevent fraud and abuse in the healthcare industry. These laws can apply to both our direct-to-consumer marketing practices as well as our marketing to
clinicians and healthcare facilities. The AKS prohibits, among other things, soliciting, offering, receiving, or paying anything of value to generate business that may be paid for, in whole or in part, by a federal healthcare program. The Civil Monetary Penalties Law covers a variety of conduct, often violations under other laws, and includes penalties for AKS violations as well as causing the submission of false claims. The False Claims Act generally prohibits anyone from knowingly and willingly presenting, or causing to be presented, any claims for payment for goods or services, including to government payors, such as Medicare and Medicaid, that are false or fraudulent including false certifications of compliance with applicable law. The federal government and states also regulate sales and marketing activities and financial interactions between manufacturers and healthcare providers and academic medical centers, requiring disclosure to government authorities and the public of such interactions, and the adoption of compliance standards or programs. State attorneys general have also taken action to regulate the marketing of prescription drugs under state consumer protection and false advertising laws.
Pricing, Reimbursement and Access Regulations. Pricing and reimbursement for our products depend in part on government regulation. Any significant efforts at the federal or state levels to reform the healthcare system by changing the way healthcare is provided or funded or to expand controls on drug pricing, implement international reference pricing, including Most-Favored-Nation (MFN) drug pricing, or impact government reimbursement and access to medicines and vaccines on public and private insurance plans or consumer purchasing platforms could have a material impact on us.
In May 2025, the Trump Administration issued an Executive Order titled “Delivering Most-Favored Nation Prescription Drug Pricing to American Patients”, outlining a plan to reduce prescription drug costs in the U.S., which was followed by formal letters to major pharmaceutical companies (including Pfizer) in July 2025 outlining steps they should take to lower prescription drug costs to U.S. patients and taxpayers (collectively, the MFN Initiatives). In September 2025, Pfizer announced an agreement with the Trump Administration in which we voluntarily agreed to implement measures designed to make certain drug prices for U.S. patients more comparable to those in other developed countries. We are also participating in the TrumpRx.gov platform, which allows U.S. patients to purchase certain medicines at significant discounts to current retail prices. The September 2025 agreement also provides a three-year grace period during which time our products will not face Section 232 tariffs, provided the Company further invests in manufacturing in the U.S. Pfizer is now in the process of entering into binding final agreements to implement these arrangements.
In addition, we must offer discounts or rebates on purchases of pharmaceutical products, and often voluntarily agree to supplemental rebates, under various government programs including Medicare, Medicaid, the Veterans Administration and the 340B Program. We also must report specific prices to state and federal government agencies. The calculations necessary to determine the prices reported are complex and the failure to do so accurately may expose us to enforcement measures.
The drug pricing provisions of the IRA have been and continue to be implemented. The IRA includes several provisions intended to lower prescription drug costs for Medicare patients and to reduce drug spending by the federal government. The IRA also made significant changes to the Medicare Part D benefit design (IRA Medicare Part D Redesign), which took effect beginning in 2025 and negatively impacted our 2025 revenues by approximately $1 billion. We do not expect a material, incremental impact from the IRA Medicare Part D Redesign in 2026 versus the baseline set in 2025. Among other things, the IRA enhanced the Medicare Part D benefit by eliminating the coverage gap (“donut hole”) beginning in 2025, added a maximum out-of-pocket cap for Medicare beneficiaries (set at $2,100 for 2026), and created a new program, the Medicare Prescription Payment Plan, that allows patients to pay their cost-sharing over time. These changes also include a new Medicare Part D Manufacturer Discount Program, which changed our discounting obligations for Medicare Part D utilization of our drugs. Specifically, this program requires manufacturers to provide a 10% discount on branded prescriptions in the initial coverage phase and a 20% discount in the catastrophic phase. The IRA also imposes rebates under Medicare Part B and Medicare Part D on drug price increases that outpace inflation, and directs HHS to set the prices of certain high-expenditure, single-source drugs and biologics covered under Medicare, known as the MDPNP. In August 2023, CMS published the first ten medicines subject to the MDPNP, which included Eliquis. In August 2024, the government released the new Medicare price for Eliquis, which, effective January 1, 2026, is required to be offered to all Medicare beneficiaries at the price established by the government (known as the Maximum Fair Price). The Maximum Fair Price also must be offered to covered entities participating in the 340B Program that dispense Eliquis to a Medicare beneficiary when the Maximum Fair Price is lower than the statutorily-mandated price such entities are offered under the 340B Program. In January 2025, CMS announced the selection of another 15 drugs from Medicare Part D for the Maximum Fair Price, with prices to be set and effective on January 1, 2027. Ibrance and Xtandi were included in the list of 15 drugs selected. Another 15 drugs from Medicare Part B or Medicare Part D were selected on January 27, 2026, for the Maximum Fair Price to be set and effective on January 1, 2028. Xeljanz was included in the list of 15 drugs selected. It is possible that more of our products could be selected in future years, which could, among other things, lead to lower revenues. The MDPNP is currently subject to legal challenges and therefore, the outcome of the MDPNP remains uncertain. We continue to evaluate the impact of the IRA on our business, operations and financial condition and results as the full effect of the IRA on our business and the pharmaceutical industry remains uncertain. See the discussion regarding the IRA in the Overview of Our Performance, Operating Environment, Strategy and Outlook—Our Operating Environment section within MD&A.
Changes to the MDRP or the 340B Program also could have a material impact on our business. For example, certain changes finalized by CMS in a December 2020 final rule, including which products qualify as “line extension” drugs subject to increased rebate liability, may have a material adverse impact on our business. Additionally, in September 2024, CMS finalized a new rule that, among other items, expands the scope of medications considered to be “covered outpatient drugs” that could be subject to rebates under the MDRP and imposes penalties on covered outpatient drugs that CMS determines to be “misclassified.” Changes to the way we calculate Average Sales Price under the Medicare Part B program, including new rules regarding the treatment of bundled sales and bona fide service fees that were finalized in a November 2025 final rule, also may impact the reimbursement amount available to providers for our Part B products administered to Medicare beneficiaries. These changes could impact our revenues for such products if Part B reimbursement amounts are negatively impacted. Many pharmaceutical manufacturers believe the 340B Program continues to expand beyond the original intent of serving low-income/uninsured patients. However, there has been limited government oversight to control this significant growth. Accordingly, several manufacturers, including Pfizer, have implemented initiatives and policies seeking to improve 340B Program integrity and transparency, such as establishing certain conditions related to use of contract pharmacies, requesting or requiring limited data submissions by covered entities, and pursuing use of rebate models.
Some of these efforts have been the subject of legal challenges and other advocacy efforts by covered entities, states, and/or the HHS Health Resources and Services Administration (HRSA), the agency that administers the 340B Program. In 2022, we implemented a policy to help improve contract pharmacy integrity. In 2021 and 2022, HRSA sent letters to numerous manufacturers (not including Pfizer) that implemented contract pharmacy policies and integrity initiatives; the letters expressed HRSA’s view that those manufacturers’ policies were in violation of the 340B Program statute. Several manufacturers challenged HRSA’s enforcement letters in federal court. We believe that our policy is consistent with the statute and supported by the federal court decisions issued to date.
In addition, some states have enacted laws seeking to address various aspects of manufacturer policies related to contract pharmacy transactions in their states. Several stakeholders have challenged such laws and litigation is ongoing in several jurisdictions. Certain courts have decided these challenges in favor of manufacturers and other courts have ruled in favor of relevant states. Additionally, other states have considered and could enact similar laws going forward, although any such laws also may be subject to legal challenges.
Additional legal or legislative developments at the federal or state level with respect to the 340B Program may have an adverse impact on our integrity initiative, and we may face enforcement action or penalties, depending upon such developments. The 340B Program continues to be a subject of congressional scrutiny and inquiries, litigation, and other developments, any or all of which could affect the scope of the 340B Program and Pfizer’s obligation to offer the 340B price to 340B Program-covered entities under the 340B Program.
States seek to control healthcare costs related to Medicaid and other state regulated healthcare programs. A majority of states use preferred drug lists to manage access to pharmaceutical products under Medicaid, including some of our products. States may seek to negotiate supplemental rebate agreements that are larger than the minimum federal requirement for preferred formulary access. Preferred access to our products under the Medicaid managed care programs are often determined by the managed care health plans contracted by the state to administer benefits, which may also require supplemental rebates for preferred formulary access. We expect states will continue to seek cost cutting, which may focus on managed care capitation payments, upper pricing limits, supplemental rebates, and/or formulary management.
Coverage and cost sharing for certain insurance programs including Medicare, Medicaid and Children’s Health Immunization Program (CHIP) can depend on recommendations from advisory committees, including the U.S. Preventive Services Taskforce (USPSTF) and the ACIP. Changes in the recommendations or structure of those committees may affect the use of our medicines and vaccines.
In the U.S., there is considerable public and government scrutiny of pharmaceutical pricing and intellectual property and we expect to see continued focus by federal and state governments on regulating pricing and access to medicine, in addition to actions already taken, which could result in legislative and regulatory changes.
Further efforts by states and the federal government to regulate prices or payment for pharmaceutical products, including proposed actions to facilitate drug importation, implement international reference pricing, including MFN drug pricing, or establish upper pricing limits that cap reimbursement to lower reference prices, require deep discounts, impose financial penalties related to pricing practices, and require manufacturers to report and make public price increases and sometimes a written justification for the increase, could adversely affect our business if implemented.
Further, commercial payors often follow Medicare coverage and reimbursement policies when setting their own payment rates. Any reduction in cost or other containment measures may similarly be adopted by commercial plans. Payors may continue to promote generic drugs and biosimilars more aggressively to generate savings and attempt to stimulate additional price competition. In addition, we expect that consolidation and integration among pharmacy chains, wholesalers and PBMs will increase pricing pressures in the industry.
Anti-Corruption. The FCPA prohibits U.S. corporations and their representatives from offering, promising, authorizing or making payments to any foreign government official, government staff member, political party or political candidate to obtain or retain business abroad. The scope of the FCPA includes interactions with certain healthcare professionals in many countries. Other countries have enacted similar anti-corruption laws and/or regulations.
Data Privacy. The number of privacy and data security laws and regulations in the U.S. to which we are subject on the federal and state level continues to increase. We routinely collect and use sensitive personal data relating to health. The legislative, regulatory and litigation landscape for privacy and data protection requirements is rapidly evolving and changing, and may limit our ability to use data globally or across borders. Data protection requirements are not universal and can conflict between jurisdictions. Compliance with those laws and regulations is made more complex by the lack of consistent standards, common definitions, or clear regulatory expectations. We also anticipate continued and new uses of data as we explore the use of AI tools both internally and externally. Enforcement of these data privacy and security laws and regulations is increasing and litigation is becoming more common, and we expect such trends will continue. Any failure or perceived failure by us to comply with applicable privacy and data protection laws and regulations, including cybersecurity breaches or incidents, could subject us to significant fines and penalties, and/or litigation, as well as negatively impact our reputation.
Outside the U.S.
New Drug Approvals. In the EU, the EMA conducts the scientific evaluation, supervision and safety monitoring of our innovative medicinal products that are eligible for the centralized marketing authorization procedure. Through the centralized procedure, pharmaceutical companies may submit to the EMA a single application for a marketing authorization valid in all the EU and the European Economic Area (EEA) countries. The EC grants marketing authorization by issuing a legally binding decision based on the EMA's recommendation. The centralized procedure is mandatory for certain new products (such as biotechnology and orphan medicine), optional for others (including new active substances and products with significant therapeutic, scientific, or technical innovation), and not available for all remaining products. In the U.K., the Medicines and Healthcare products Regulatory Agency (MHRA) is the sole regulatory authority, and companies must obtain an MHRA marketing authorization to market medicines in the U.K. In Japan, the Pharmaceuticals and Medical Device Agency is involved in a wide range of regulatory activities, including clinical studies, approvals, post-marketing reviews and pharmaceutical safety. In China, the National Medical Product Administration is the primary regulatory authority for approving and supervising medicines. Health authorities in many middle- and lower-income countries might require marketing approval or scientific opinions by a recognized regulatory authority (e.g., the FDA or EMA/EC) before they begin reviewing or approving applications. By way of example, the EMA, in cooperation with the WHO, can provide scientific opinions on high priority human medicines, including vaccines, for markets outside the EU.
In the EU, the European Council and the European Parliament reached an agreement in December 2025 on the most significant reform of the EU’s Pharmaceutical Legislation in 20 years (the EU Pharma Package). The reform still requires formal approval by both institutions and will enter into force upon publication in the EU’s Official Journal, expected in 2026. The reform encompasses a broad range of measures, including changes to regulatory exclusivity, incentives to combat microbial resistance, intellectual property exemptions for generic medicines, and marketing authorization procedures. Most provisions are expected to apply from 2028, following a two-year transition period. This landmark reform is expected to significantly influence the way innovative medicines are developed, authorized, monitored, and accessed across the EU. Once implemented, certain provisions of the reform could potentially have an adverse impact on our business.
In the EU, several other recently adopted or proposed legislative initiatives may affect our business, including the EU HTA-R, new rules on
substances of human origin (SoHO Regulation), the ongoing reform of the rules governing SPCs, the EU Critical Medicines Act, and the EU Biotech Act. In addition, a number of EU regulations that are not primarily focused on medicinal products may nevertheless have a material impact on pharmaceutical companies. These include the EU AI Act, EU Data Act, and the EU Health Data Space Regulation, among others. Moreover, the EU is currently discussing the Digital Omnibus Package, which comprises a series of technical amendments to a wide body of EU digital legislation.
Pharmacovigilance. In the EU, the EMA’s PRAC is responsible for reviewing and making recommendations on product safety issues. Specifically, the PRAC focuses on detecting, assessing and communicating the risks associated with adverse reactions of medicinal products, while considering their therapeutic effects. It also evaluates post-authorization safety studies and conducts pharmacovigilance audits. Outside developed markets, pharmacovigilance requirements vary and are generally not as extensive, but there is a trend toward increasing regulation.
Pricing and Reimbursement. Certain governments, including in the different EU member states, the U.K., Japan, China, Canada and Australia provide healthcare at low-to-zero direct cost to consumers at the point of care and have significant power to regulate pharmaceutical prices or patient reimbursement levels to control costs for the government-sponsored healthcare system, particularly under global financing pressures. Governments globally may use a variety of measures to control costs, including, among others, legislative or regulatory pricing reforms, cross country collaboration and procurement, price cuts, mandatory rebates, health technology assessments, forced localization as a condition of market access, international reference pricing (i.e., the practice of a country linking its regulated medicine prices to those of other countries), QCE processes and VBP. In addition, the international patchwork of price regulation, differing economic conditions and incomplete value assessments across countries has led to varying access to quality medicines in many markets and some third-party trade in our products between countries.
In China, pricing pressures have increased in recent years because of an overall focus on healthcare cost containment with the central government emphasizing improved health outcomes and decreased drug prices as key indicators of progress towards its healthcare reform. State owned hospitals and the state insurance program account for the vast majority of all drug purchases, despite recent government efforts to promote use of commercial insurance for some innovative products. For patented innovative products, drug prices have decreased dramatically as a result of adding innovative drugs (including oncology medicines, pediatric medicines, orphan drugs and medicines for chronic diseases) to the National Reimbursement Drug List via access-price negotiation. Certain patented innovative drug products are also listed for enrollment into the Catalogue of Innovative Drugs for Commercial Insurance and subject to a similar price negotiation process with selected commercial insurance stakeholders. A centralized VBP program with a tendering process implemented at both the national level and the provincial level aims to contain healthcare costs by driving utilization of generics that have passed QCE. This has resulted in further lowering the price of medicines, especially off-patent medicines; this trend is expected to continue. China is continuing its use of Health Technology Assessment, a tool used to evaluate clinical effectiveness and economic value to manage public health budgets, and is controlling mark-ups within the country using a two-invoice limited system, aiming to regulate the pricing of pharmaceutical products and certain types of medical devices. Pfizer, along with most off-patent originators, has mostly not been successful in the VBP bidding process. The government has indicated that additional drugs which are past patent-based exclusivity expiry (including biological products) could be subjected to VBP qualification in future rounds. Pfizer did not participate in the eleventh national VBP round in 2025 because it did not include any relevant Pfizer products. While certain details of future QCE expansion have been made available, we are unable to determine the impact on our business of the various pricing measures underway.
Healthcare Provider Transparency and Disclosures. Several countries have implemented laws requiring (or industry trade associations have recommended) disclosure of transfers of value made by pharmaceutical companies to healthcare providers and/or healthcare organizations, such as academic teaching hospitals.
Intellectual Property. Reliable patent protection and enforcement around the world are among the key factors we consider for continued business and R&D investment. The WTO Agreement on Trade-Related Aspects of Intellectual Property Rights (WTO-TRIPS) requires participant countries to provide patent and other intellectual property-related protection for pharmaceutical products by law, with a time-limited exemption provided for least-developed countries. While some countries have made improvements, we still face patent grant, enforcement and other intellectual property challenges in many countries.
While the global intellectual property policy environment has generally improved following implementation of WTO-TRIPS and bilateral/multilateral trade agreements, our growth and ability to bring new product innovation to patients depends on maintaining those standards and further progress in intellectual property protection. In certain developed international markets, governments maintain relatively effective intellectual property policies. However, in the EU, the review of the pharmaceutical legislation is reducing or modifying the overall period of regulatory data and market protection. In several emerging market countries and multilateral institutions, governments continue to address the role of intellectual property in the context of, for example, access to medicines.
Considerable political and economic pressure has weakened current intellectual property protection in some countries and has led to policies such as more restrictive standards for obtaining patents and more difficult procedures for patenting biopharmaceutical inventions, restrictions on patenting certain types of inventions, revocation of patents, laws or regulations that promote or provide broad discretion to issue a compulsory license, weak intellectual property enforcement and failure to implement effective regulatory data protection. Our industry advocacy efforts focus on seeking a fair and transparent business environment for foreign manufacturers, underscoring the importance of strong intellectual property systems for all innovative industries (both domestic and foreign) and helping improve patients’ access to innovative medicines and vaccines.
Data Privacy. We are subject to extensive privacy and data protection laws and regulations around the world concerning the collection, use and sharing of personal data. We routinely collect and use sensitive personal data relating to health. The legislative, regulatory and litigation landscape for privacy and data protection requirements is rapidly evolving and changing, and may limit our ability to use data globally or across borders. Data protection requirements are not universal and can conflict between jurisdictions. Compliance with those laws and regulations is made more complex by the lack of consistent standards, common definitions, or clear regulatory expectations. We also anticipate continued and new uses of data as we explore the use of AI tools both internally and externally. Enforcement of these data privacy and security laws and regulations is increasing and litigation is becoming more common, and we expect such trends will continue. Any failure or perceived failure by us to comply with applicable privacy and data protection laws and regulations, including cybersecurity breaches or incidents, could subject us to significant fines and penalties, and/or litigation, as well as negatively impact our reputation.
ENVIRONMENTAL MATTERS
Our operations are affected by national, state and/or local environmental laws. We have made, and intend to continue to make, the expenditures necessary for compliance with applicable laws. We also are cleaning up environmental contamination from past industrial activity at certain sites. We incurred capital and operational expenditures in 2025 for environmental compliance purposes and for the clean-up of certain past industrial
activity as follows: approximately $125 million in environment-related capital expenditures and approximately $188 million in other environment-related expenses.
While capital expenditures or operating costs for environmental compliance cannot be predicted with certainty, we do not currently anticipate they will have a material effect on our capital expenditures or financial position.
As a science guided organization, we take a proactive approach to our environmental sustainability initiatives. In 2022, we announced a goal to further reduce greenhouse gas (GHG) emissions and achieve the Science Based Target Initiative’s voluntary Net-Zero Standard by 2040. As part of this goal, Pfizer aims to decrease its GHG emissions by 95% and its value chain emissions by 90% from 2019 levels by 2040. To support our goal, we are developing and implementing our emission reduction plans, including strategies to achieve reductions throughout our value chain and setting expectations for our suppliers to establish science-aligned GHG emission reduction goals. Our emission reduction plan-related expenses and capital spending incurred for 2025 were not material to our consolidated financial statements. While we expect to incur incremental capital and operational expenditures to meet our goal, we do not currently anticipate they will have a material effect on our financial position in the near term. Longer term uncertainties such as the likelihood of commercially available technologies make it difficult to predict the financial impact of meeting the goal. We continue to assess and monitor the financial impact of our GHG emission reduction plans.
OUR PEOPLE
Our purpose is: Breakthroughs that change patients’ lives. These breakthroughs are delivered through the collaboration of our talented workforce. As of December 31, 2025, we employed approximately 75,000 people worldwide. Our ability to successfully deliver on our purpose is dependent on our people. Creating a purpose-driven workplace that attracts, nurtures, and retains top talent is a priority. We strive to build a vibrant and supportive work culture that is designed to empower colleagues to innovate, collaborate, and contribute meaningfully to improving global health. We invest in comprehensive development programs, provide merit-based opportunities for advancement, and encourage work-life integration through flexible work arrangements. We work to create an environment that prioritizes colleagues’ health and wellness. Our people-centric approach touches every aspect of the employee experience — including recruiting, benefits, compensation and development. Our Pfizer values — courage, excellence, equity and joy — remain core to all that we do.
Culture. We cultivate community in our workplace to deliver on our purpose. As we work to bring together people with different perspectives and experiences we strive to foster a collaborative environment. We continue to execute a merit-based talent approach, focusing on identifying candidates with the right qualifications and ensuring they are considered for the opportunities based on their skills, abilities, and performance. We aim to provide everyone with an opportunity to demonstrate their merit. Our leaders set the tone for the company, embracing accountability and transparency, while promoting a vibrant culture in which colleagues are free to speak up and are encouraged to share views and raise concerns without fear of retaliation.
Colleague Engagement. We are committed to helping our colleagues reach their full potential by recognizing their performance and leadership capabilities, while providing meaningful opportunities for growth and development. Open communication and feedback are foundational to performance, colleague engagement, and teamwork. Pfizer managers regularly engage in discussions on colleague performance and leadership behaviors to encourage breakthrough goals and strengthen leadership capabilities. Equally important is our commitment to listening and responding to colleague feedback, fostering a healthy work environment that attracts and retains talent.
We are passionate about creating safe spaces at work, so our colleagues feel able and encouraged to provide feedback and raise concerns and questions. The Office of the Ombuds provides information and guidance to help colleagues address and resolve work-related issues. We also host company-wide safe space calls and provide various other public, private, and anonymous channels for colleagues to speak up without fear of retaliation.
We gather colleague feedback through multiple channels to help ensure a comprehensive understanding of colleague experiences and needs, including an annual engagement survey that helps us track priority areas and equip leaders with actionable insights. Additionally, we conduct focus groups, check-in surveys, and colleague forums at various points in the employee lifecycle. These additional listening mechanisms are designed to capture real-time feedback, adapt to evolving needs, and continuously improve our ways of working.
Colleague recognition drives engagement, a sense of belonging, motivation, and productivity. Our global rewards and recognition program, Bravo, lets colleagues celebrate and acknowledge each other for demonstrating Pfizer values in a way that makes an impact on the company, a colleague, a team or a patient.
Performance and Leadership. Pfizer prioritizes a leadership mindset which complements our Pfizer values and behaviors and is designed to foster a workplace that is more dynamic, innovative, and compassionate, empowering all team members to contribute to our ambitious business goals. Our “Project-Based Ways of Working” allows colleagues to take on leadership roles and decision responsibilities through training, coaching, and a robust support network. Our performance management approach provides our colleagues and their managers with opportunities to set goals, receive feedback and engage in discussions on performance aimed at helping colleagues grow and develop.
Growth and Development. Colleague growth is at the heart of building a future-ready workforce that thrives amid change and drives innovation, while also empowering each colleague to achieve their personal aspirations and success. As we navigate an evolving healthcare landscape, staying agile and competitive is essential to delivering breakthroughs for patients. That is why we prioritize opportunities for learning, skill-building, and growth. Such opportunities are designed to equip colleagues with the skills to tackle new challenges and seize emerging opportunities, including related to AI and technology innovation. Investing in career development not only reaffirms our commitment to colleagues’ potential, but also drives engagement, productivity, and overall job satisfaction.
Health, Safety and Well-Being. At Pfizer, protecting the health, safety, and well-being of colleagues and contingent workers, all of whom are essential to driving our business forward, is an integral part of how we operate. Pfizer's Global Environment, Health & Safety (EHS) Policy and supporting standards outline our approach to assessment, evaluation, elimination, and mitigation of EHS risks across our operations globally. Our leadership is accountable for EHS compliance and risk management. Colleagues and contingent workers receive EHS training relevant to their job roles, including measures to prevent workplace incidents and injuries. In addition, we conduct workplace assessments, inspections, and audits to evaluate and continuously improve our EHS arrangements. We are committed to supporting and encouraging our colleagues’ well-being. We use results from our annual colleague engagement survey and other colleague feedback forums to inform the wellness services we offer.
Pay Equity. Our commitment to pay equity for all colleagues is based on our values and our intention to continue to build a highly motivated workforce. We are committed to equitable pay practices at Pfizer for colleagues based on role, education, experience, performance, and location and we conduct a global pay equity analysis on an annual basis.
| No annotations |
A subscription is required for the below company data including historic share prices, economic, EV bridge, WACC, customizable comparables and earnings forecasts.
Sign up today or log in to your account| 52 week | high $97.88 | low $68.39 |
| Economic Data | |
| Historic real GDP growth rate | ?? |
|---|---|
| Forecast real GDP growth rate | ?? |
| Historic inflation rate | ?? |
| Forecast inflation rate | ?? |
| SOFR | ?? |
| US 3 month Treasury Yield | ?? |
| Calculating WACC | ||
| Govt. bond yield* | ?? | |
|---|---|---|
| Eq. risk premium* | ?? | |
| Adj. beta | ?? | |
| Est. cost of equity | ||
| Unlevered beta | ?? | |
| Credit spread* | ?? | |
| Cost of debt | ||
| Credit rating (Moodys/S&P) | ?? | |
| Total debt / capitalization | ?? | |
| Total debt / mkt. cap. | ?? | |
| WACC | ||
| Earnings | |||||
|---|---|---|---|---|---|
| Date | ?? | ?? | ?? | ||
| Revenue ($M) | ?? | ?? | ?? | ||
| Growth | ?? | ?? | ?? | ||
| EBITDA ($m) | ?? | ?? | ?? | ||
| Margin | ?? | ?? | ?? | ||
| EPS ($) | ?? | ?? | ?? | ||
Market data by Finnhub | Earnings forecasts by Factset | Economic data from FRED |
Fundamental data by Financial Edge
Please notify us of any data inaccuracies. All critical data usage must be externally validated.
Please notify us of any data inaccuracies. All critical data usage must be externally validated.